


ABSTRACT
Background and Aim Brucellosis is a globally significant zoonotic disease that causes reproductive failure in animals and chronic infection in humans, resulting in substantial economic and public health burdens. The pathogenicity of Brucella species is largely mediated by virulence determinants such as the type IV secretion system and lipopolysaccharide biosynthetic machinery. Despite extensive studies on individual components, a comparative and integrative understanding of conserved virulence factors across major Brucella species remains limited. This study aimed to perform a comprehensive in silico characterization of key virulence proteins across representative Brucella species to identify potential targets for vaccine and antimicrobial development.
Materials and Methods: Nine virulence-associated proteins (VirB3, VirB5, VirB7, WbkA, WbkB, WbkC, FabZ, gmd, and lpxC) from five major Brucella species were analyzed using bioinformatics approaches. Analyses included physicochemical characterization, subcellular localization prediction, conserved domain and motif identification, multiple sequence alignment, phylogenetic analysis, and protein–protein interaction network construction. Publicly available databases and tools such as NCBI, ProtParam, DeepLoc, MEME, MEGA11, and STRING were utilized.
Results: Subcellular localization analysis revealed that VirB5 is extracellular and VirB7 is outer membrane-associated, whereas most other proteins were cytoplasmic or membrane-associated. Conserved motif analysis identified three shared motifs, particularly in VirB5 and WbkB, indicating functional conservation across species. Phylogenetic and sequence alignment analyses demonstrated high conservation of virulence proteins among the selected Brucella species. Protein-protein interaction networks highlighted VirB3, VirB5, VirB7, WbkC, FabZ, gmd, and lpxC as key interaction hubs. lpxC showed strong connectivity with lipid A biosynthesis proteins, suggesting its central functional role.
Conclusion This integrative in silico analysis identified conserved virulence proteins with potential translational relevance. VirB5 and VirB7 emerged as promising candidates for subunit vaccine development due to their extracellular or membrane localization and conserved motifs, while lpxC was identified as a potential antimicrobial target because of its central role in lipopolysaccharide biosynthesis. These findings provide a rational framework for future experimental validation and support the development of improved control strategies against brucellosis.
Keywords: bioinformatics, Brucella, lipopolysaccharide, lpxC, protein–protein interaction, type IV secretion system, vaccine targets, virulence factors.
INTRODUCTION
Brucellosis, also referred to as Mediterranean fever, Cyprus fever, Malta fever, and undulant fever [1], is one of the most economically important and widely distributed bacterial zoonoses worldwide [1–3]. The causative agents primarily colonize the reproductive system of domestic animals, particularly Brucella abortus and Brucella melitensis, and can lead to chronic, sometimes lifelong, debilitating infections in humans. These infections are characterized by severe clinical manifestations, including arthritis, fever, splenomegaly, and hepatomegaly [2].
The disease affects domestic animals such as cattle, goats, sheep, and pigs, causing abortion and metritis in females, and orchiepididymitis in males, ultimately leading to infertility, reduced fertility, and decreased milk production. Humans act as accidental hosts. Transmission occurs through direct contact with contaminated materials such as placenta, vaginal discharge, or aborted fetuses [1], inhalation of airborne bacteria, or ingestion of contaminated dairy products. Human-to-human transmission, although rare, may occur through organ transplantation, blood transfusion, or breastfeeding [4].
Bacteria of the genus Brucella belong to the phylum Proteobacteria, class Alphaproteobacteria, order Rhizobiales, and family Brucellaceae [1, 5]. The genus includes major pathogenic species such as Brucella suis, B. melitensis, and B. abortus, which are responsible for significant economic losses in livestock and are also associated with human infections [4]. Additional identified species include Brucella canis, Brucella ovis, Brucella pinnipedialis, Brucella neotomae, Brucella microti, Brucella ceti, Brucella papionis, Brucella inopinata, and Brucella vulpis [4, 5]. These organisms are non-spore-forming, non-encapsulated, non-motile, Gram-negative coccobacilli or short bacilli, and are facultative intracellular pathogens [6]. They possess the ability to adapt to the macrophage environment, survive, and replicate, as well as tolerate acidic pH and low oxygen conditions [1].
The genomes of all Brucella species exhibit similar size and structure, with an average genome size of approximately 3.29 Mb, consisting of two circular chromosomes. Chromosome I is approximately 2.11 Mb, while chromosome II is around 1.18 Mb, with G+C contents of 57.2% and 57.3%, respectively [6]. A major virulence factor of Brucella is lipopolysaccharide (LPS), which comprises lipid A, a core oligosaccharide, and an O-antigen chain [7]. Wild-type strains such as B. melitensis, B. suis, B. abortus, B. neotomae, B. microti, B. ceti, and B. pinnipedialis express smooth (S-type) LPS, whereas B. ovis and B. canis lack the O-polysaccharide and exhibit rough (R-type) LPS [8].
The lipid A component anchors LPS within the cell membrane, while the O-polysaccharide extends outward, determining the immunological properties of the bacterium. Diagnosis of brucellosis is based on detecting antibodies against the O-polysaccharide in human and animal sera [9]. LPS plays a crucial role in intracellular survival, exhibiting low endotoxicity, resistance to macrophage degradation, and evasion of host immune responses [10].
A total of nineteen genes essential for LPS synthesis and smooth phenotype expression have been identified in B. melitensis [11]. Most LPS biosynthesis genes are clustered in the wbo and wbk regions, with the wbk locus serving as the primary genetic region for O-polysaccharide synthesis. This region encodes enzymes involved in N-formylperosamine synthesis (per, gmd, wbkC genes), O-polysaccharide glycosyltransferases (wbkA, wbkE genes), ABC transporters (wzt, wzm genes), polyisoprenyl-phosphate N-acetylhexosamine-1-phosphate transferase (wbkF gene), and enzymes required for N-acetylamino sugar synthesis (wbkD gene) [12].
β-hydroxyacyl-acyl carrier protein dehydratase (FabZ) plays a key role in fatty acid biosynthesis during elongation cycles [13] and has also been associated with LPS formation [14]. The gene products of per, gmd, and wbkC are predicted to participate in 4-formamido-4,6-dideoxymannose synthesis [15]. Experimental studies have demonstrated that wbkC is essential for O-side-chain production, while wbkA is likely involved in O-side-chain polymerization due to its similarity to mannosyltransferases. The LPS structure of Brucella differs significantly from that of other Enterobacteriaceae such as Escherichia coli [15, 16], further contributing to its role as a virulence factor [17]. Genes involved in LPS synthesis, including lpsA, lpsB/lpcC, lpxA, lpxB, lpxC, lpxD, lpxE, gmd, per, wbkA, wbkB, wbkC, wbpL, wbdA, wzm, and wzt, are unique to Brucella isolates [18].
The type IV secretion system (T4SS), first identified in B. suis and encoded by the VirB operon, is highly conserved across all Brucella species. This system consists of twelve genes (virB1–virB12) responsible for secreting bacterial macromolecules [19, 20]. The expression of the VirB operon is regulated by the quorum-sensing regulator vjbR [21]. These proteins are essential for intracellular survival, enabling bacteria to establish a replication niche by interacting with the endoplasmic reticulum and acquiring its membrane [5]. The system is rapidly activated during intracellular infection, reaching peak activity within five hours and subsequently declining after the formation of replication vacuoles [19].
Despite extensive investigations into Brucella virulence mechanisms, most previous studies have focused on individual virulence factors or single species, resulting in a fragmented understanding of pathogenicity across the genus. In particular, key components such as the type IV secretion system and LPS biosynthesis pathways have been studied in isolation, with limited efforts to integrate their structural, functional, and interaction-based characteristics across multiple clinically relevant Brucella species. Furthermore, although several virulence-associated genes have been identified, their comparative conservation, subcellular localization, motif architecture, and interaction networks remain insufficiently explored in a unified analytical framework. This lack of integrative analysis restricts the ability to prioritize robust and broadly conserved targets for vaccine or antimicrobial development.
In addition, current vaccine strategies against brucellosis remain suboptimal, with limitations in cross-protection, safety, and differentiation of infected from vaccinated animals. Similarly, the identification of novel antimicrobial targets is hindered by an incomplete understanding of essential proteins within interconnected virulence pathways. Although bioinformatics tools are widely available, their combined application for comparative prioritization of conserved virulence determinants across multiple Brucella species has not been comprehensively addressed. Therefore, a systematic, multi-dimensional in silico evaluation of conserved virulence proteins is needed to bridge this gap and support rational target selection for translational research.
The present study was designed to address this gap by performing a comprehensive and integrative in silico characterization of selected virulence-associated proteins across major Brucella species. Specifically, the study aimed to comparatively analyze key proteins involved in the type IV secretion system and LPS biosynthesis, including VirB3, VirB5, VirB7, WbkA, WbkB, WbkC, FabZ, gmd, and lpxC, using multiple complementary bioinformatics approaches.
The objectives were to evaluate their physicochemical properties, predict subcellular localization, identify conserved domains and motifs, assess evolutionary relationships through phylogenetic analysis, and construct protein–protein interaction networks to determine functional connectivity. By integrating these analytical dimensions, the study sought to identify conserved and functionally significant proteins with potential translational relevance.
Ultimately, this work aims to provide a rational framework for prioritizing candidate proteins for subunit vaccine development and antimicrobial targeting, contributing to improved control strategies against brucellosis from a One Health perspective.
MATERIALS AND METHODS
Ethical approval
This study was conducted exclusively using publicly available genomic and proteomic datasets retrieved from the National Center for Biotechnology Information (NCBI) database and other open-access bioinformatics resources. No live animals, human participants, or biological samples were involved at any stage of the study. Therefore, ethical approval from an institutional animal care and use committee or human ethics committee was not required.
Study period and location
The study was conducted using publicly available genomic and proteomic data retrieved from the NCBI database. All analyses were performed in silico between April 2024 and February 2026.
Selection of Brucella species and virulence genes
The study design and analytical workflow are presented in Figure 1. The selection of Brucella species in this study was guided by biological, epidemiological, and phenotypic diversity considerations rather than random inclusion. The five selected species were chosen based on their established relevance to animal health and, in several cases, human infections, as well as their representation across multiple host species, including cattle, sheep, goats, pigs, and dogs.
Furthermore, both smooth and rough phenotypes were intentionally included to reflect structural variability in LPS composition and the associated immunological differences. This strategy ensured a broader representation of biological and epidemiological diversity across major Brucella lineages. The inclusion of both phenotypes also enhanced diversity in LPS structures, which are known to influence immune recognition, virulence, and serological behavior.
In this study, the objective was to explore conserved molecules with potential relevance as vaccine or therapeutic targets across B. melitensis bv. 1 str. 16M, B. suis 1330, B. canis ATCC 23365, B. ovis ATCC 25840, and B. abortus 2308.
The selection of nine virulence-associated genes (virB3, virB7, virB5, wbkC, wbkB, wbkA, fabZ, gmd, and lpxC) was based on their functional roles in key pathogenic mechanisms. In addition, the availability of sequence data for all selected species in open-access databases was considered as an important methodological criterion. These proteins were selected due to their established involvement in virulence, LPS biosynthesis, and host-pathogen interactions.
Figure 1. Schematic representation of the study design and analytical workflow.
Retrieval of gene and protein sequences
The genes (virB3, virB7, virB5, wbkC, wbkB, wbkA, fabZ, gmd, and lpxC) and their corresponding amino acid sequences from B. melitensis bv. 1 str. 16M, B. suis 1330, B. canis ATCC 23365, B. ovis ATCC 25840, and B. abortus 2308 were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/gene/) (Accessed April 17, 2024).
Physicochemical characterization of virulence proteins
The physicochemical properties of the nine selected virulence proteins were analyzed using the ProtParam tool available on the ExPASy server (http://web.expasy.org/protparam/) (Accessed April 17, 2024). The parameters evaluated included the number of amino acids, molecular weight, theoretical isoelectric point, instability index, and the grand average of hydropathicity (GRAVY).
Prediction of subcellular localization
The predicted subcellular localization of the virulence proteins was determined using the DeepLoc-1.0 online server (https://services.healthtech.dtu.dk/services/DeepLoc-1.0/) (Accessed April 18, 2024). Protein sequences were submitted in FASTA format for analysis.
Conserved domain identification and motif analysis
Information regarding conserved domains of the virulence proteins was obtained from the Conserved Domains Database (CDD) at NCBI (https://www.ncbi.nlm.nih.gov/guide/domains-structures/) (Accessed June 29, 2024). Protein sequences in FASTA format were submitted and analyzed at an E-value threshold of 0.01. The output displayed the best-scoring domain model along with its associated domain superfamily for each query region.
Conserved motif identification was performed using the Multiple Em for Motif Elicitation (MEME) server (Version 5.5.9) (https://meme-suite.org/meme/tools/meme) (Accessed Feb 19, 2026) for VirB3, VirB7, VirB5, WbkC, WbkB, WbkA, FabZ, gmd, and lpxC proteins.
Multiple sequence alignment and phylogenetic analysis
Multiple sequence alignments were conducted using the amino acid sequences of the selected virulence proteins in Geneious Prime version 2024.0 (Biomatters; https://www.geneious.com). These analyses were used to evaluate the degree of similarity and divergence among virulence proteins.
Phylogenetic relationships were inferred using the Maximum Likelihood method with the Jones–Taylor–Thornton (JTT) matrix-based model [22]. The dataset consisted of 24 amino acid sequences, and evolutionary analyses were performed using Molecular Evolutionary Genetics Analysis (MEGA11).
Protein–protein interaction network analysis
Protein–protein interaction networks were constructed using the STRING database (https://string-db.org/cgi/input?sessionId=bgzpUl8u2HmH) (Accessed May 03, 2024). The proteins VirB3, WbkB, WbkC, FabZ, gmd, and lpxC from B. abortus 2308 were selected to identify their interaction partners and functional connectivity.
Software versions
The bioinformatics tools and software resources used in this study, along with their respective versions and access details, are summarized in Table 1.
Table 1. Bioinformatics tools and software resources used in the study.
| Bioinformatic | Software name | Software version/ Accessed year | Software link |
|---|---|---|---|
| Physicochemical properties | Expasy Server | 2024 | http://web.expasy.org/protparam/ |
| Prediction subcellular localization | DeepLoc-1.0 | 2024 | https://services.healthtech.dtu.dk/services/DeepLoc-1.0/ |
| Conserved domain | Conserved Domains NCBI | 2024 | https://www.ncbi.nlm.nih.gov/guide/domains-structures/ |
| Motif analysis | MEME server | Version 5.5.9 | https://meme-suite.org/meme/tools/meme |
| Multiple sequence alignments | Geneious Prime | Version 2024.0 | https://www.geneious.com |
| Phylogenetic analysis | Molecular Evolutionary Genetics Analysis (MEGA) | MEGA11 | https://www.megasoftware.net/ |
| Protein–protein interaction network analysis | STRING | 2024 | https://string-db.org/cgi/input?sessionId=bgzpUl8u2HmH |
RESULTS
Retrieval of gene and protein sequences
Gene and protein sequence information for B. melitensis bv. 1 str. 16M, B. suis 1330, B. canis ATCC 23365, B. ovis ATCC 25840, and B. abortus 2308 were obtained from the NCBI Gene database. The selected virulence-associated genes included virB3, virB7, virB5, wbkC, wbkB, wbkA, fabZ, gmd, and lpxC, together with the amino acid sequences of the proteins they encode. For each gene and its corresponding protein, key annotation details such as Gene ID, gene symbol, functional description, gene type, protein accession number (NCBI RefSeq), chromosomal location, and genomic reference sequence were compiled and organized. These features are summarized in Table 2 and served as the reference dataset for the subsequent motif, localization, phylogenetic, and protein–protein interaction analyses.
Table 2. The gene and protein features of virulence genes family members in Brucellaceae.
| Species | Gene ID | Gene symbol | Gene description | Gene type | Protein accession (NCBI reference sequence) | Protein symbol | Chr. No. | Genomic sequence |
|---|---|---|---|---|---|---|---|---|
| B. melitensis bv. 1 str. 16M | 29595902 | BME_RS10325 | type IV secretion system protein VirB3 | protein coding | WP_002966512.1 | VirB3 | II | NC_003318.1 |
| B. suis 1330 | 45053164 | BR_RS10375 | NC_004311.2 | |||||
| B. canis ATCC 23365 | 55591804 | BCAN_RS10430 | NC_010104.1 | |||||
| B. ovis ATCC 25840 | 45125486 | BOV_RS10650 | NC_009504.1 | |||||
| B. abortus 2308 | 3827980 | BAB_RS26675 | NC_007624.1 | |||||
| B. melitensis bv. 1 str. 16M | 29595028 | BME_RS10345 | lipoprotein | protein coding | WP_002966516.1 | VirB7 | II | NC_003318.1 |
| B. suis 1330 | 45053160 | BR_RS10355 | NC_004311.2 | |||||
| B. canis ATCC 23365 | 55591800 | BCAN_RS10410 | NC_010104.1 | |||||
| B. ovis ATCC 25840 | 45125482 | BOV_RS10630 | NC_009504.1 | |||||
| B. abortus 2308 | NC_007624.1 | BAB_RS26655 | NC_007624.1 | |||||
| B. melitensis bv. 1 str. 16M | 29595513 | virB5 | P-type DNA transfer protein VirB5 | protein coding | WP_002966514.1 | VirB5 | II | NC_003318.1 |
| B. suis 1330 | 45053162 | WP_006191582.1 | NC_004311.2 | |||||
| B. canis ATCC 23365 | 55591802 | WP_004692616.1 | NC_010104.1 | |||||
| B. ovis ATCC 25840 | 45125484 | WP_006015000.1 | NC_010104.1 | |||||
| B. abortus 2308 | NC_007624.1 | WP_006077544.1 | NC_007624.1 | |||||
| B. melitensis bv. 1 str. 16M | 29594278 | wbkC | LPS biosynthesis N-formyltransferase WbkC | protein coding | WP_002963675.1 | WbkC | I | NC_003317.1 |
| B. canis ATCC 23365 | 55590265 | NC_010103.1 | ||||||
| B. ovis ATCC 25840 | 45123991 | NC_009505.1 | ||||||
| B. abortus 2308 | 3787272 | NC_007618.1 | ||||||
| B. suis 1330 | 45051620 | WP_006189959.1 | NC_004310.3 | |||||
| B. melitensis bv. 1 str. 16M | 29594264 | wbkB | protein WbkB | protein coding | WP_002963676.1 | WbkB | I | NC_003317.1 |
| B. suis 1330 | 45051621 | WP_004691951.1 | NC_004310.3 | |||||
| B. canis ATCC 23365 | 55590266 | WP_004691951.1 | NC_010103.1 | |||||
| B. ovis ATCC 25840 | 45123992 | WP_006011447.1 | NC_009505.1 | |||||
| B. abortus 2308 | 3787273 | WP_002963676.1 | NC_007618.1 | |||||
| B. melitensis bv. 1 str. 16M | 29594259 | wbkA | family 1 glycosyltransferase WbkA | protein coding | WP_006642693.1 | WbkA | I | NC_003317.1 |
| B. suis 1330 | 45051629 | NC_004310.3 | ||||||
| B. canis ATCC 23365 | 55590273 | NC_010103.1 | ||||||
| B. ovis ATCC 25840 | 45124000 | WP_280176887.1 | NC_009505.1 | |||||
| B. abortus 2308 | 3787283 | WP_002969840.1 | C_007618.1 | |||||
| B. melitensis bv. 1 str. 16M | 29593646 | fabZ | 3-hydroxyacyl-ACP dehydratase FabZ | protein coding | WP_004683864.1 | FabZ | I | NC_003317.1 |
| B. ovis ATCC 25840 | 45124527 | WP_004688426.1 | NC_009505.1 | |||||
| B. suis 1330 | 45052194 | NC_004310.3 | ||||||
| B. canis ATCC 23365 | 55590834 | NC_010103.1 | ||||||
| B. abortus 2308 | 3788700 | WP_002964280.1 | NC_007618.1 | |||||
| B. melitensis bv. 1 str. 16M | 29596094 | gmd | GDP-mannose 4,6-dehydratase | protein coding | WP_004681750.1 | gmd | II | NC_003318.1 |
| B. suis 1330 | 45053472 | WP_004687379.1 | NC_004311.2 | |||||
| B. canis ATCC 23365 | 5592109 | WP_004687379.1 | NC_010104.1 | |||||
| B. ovis ATCC 25840 | 45125769 | WP_006015606.1 | NC_009504.1 | |||||
| B. abortus 2308 | 3788735 | WP_002963680.1 | NC_007618.1 | |||||
| B. melitensis bv. 1 str. 16M | 29593377 | lpxC | UDP-3-O-acyl-N-acetylglucosamine deacetylase | protein coding | WP_004684016.1 | lpxC | I | NC_003317.1 |
| B. suis 1330 | 45052435 | WP_002964532.1 | NC_004310.3 | |||||
| B. canis ATCC 23365 | 55591076 | NC_010103.1 | ||||||
| B. ovis ATCC 25840 | 45124767 | NC_009505.1 | ||||||
| B. abortus 2308 | 3788783 | NC_007618.1 |
Physicochemical properties of proteins
The physicochemical properties of nine virulence proteins were predicted and analyzed via using PortParam to obtain their number of amino acids, molecular weight, theoretical isoelectric point, instability coefficient, and the grand average of hydropathicity (GRAVY) (Table 3).
Table 3. Predicting results of the physicochemical properties of virulence proteins.
| Protein accession (NCBI Reference sequence) | Number of aa | Mw (Da) | pI | Instability index | GRAVY |
|---|---|---|---|---|---|
| WP_002966512.1 (VirB3) | 116 | 13081.61 | 11.41 | 36.22 (stable) | 0.309 (hydrophobic) |
| WP_002966516.1(VirB7) | 57 | 5931.01 | 8.98 | 44.71 (unstable) | 0.182 (hydrophobic) |
| WP_002966514.1 (VirB5) | 238 | 26810.35 | 5.61 | 48.87 (unstable) | –0.599 (hydrophilic) |
| WP_006191582.1 (VirB5) | 238 | 26860.41 | 5.47 | 48.72 (unstable) | –0.589 (hydrophilic) |
| WP_004692616.1 (VirB5) | 238 | 26811.33 | 5.47 | 49.43 (unstable) | –0.599 (hydrophilic) |
| WP_006015000.1 (VirB5) | 238 | 26800.31 | 5.61 | 49.68 (unstable) | –0.595 (hydrophilic) |
| WP_006077544.1 (VirB5) | 238 | 26829.40 | 5.67 | 49.68 (unstable) | –0.604 (hydrophilic) |
| WP_002963675.1 (WbkC) | 259 | 29128.48 | 5.89 | 38.31 (stable) | –0.033 (hydrophilic) |
| WP_006189959.1 (WbkC) | 259 | 29110.45 | 5.89 | 38.60 (stable) | –0.023 (hydrophilic) |
| WP_002963676.1 (WbkB) | 284 | 32322.82 | 5.52 | 49.07 (unstable) | –0.119 (hydrophilic) |
| WP_004691951.1 (WbkB) | 284 | 32381.93 | 5.72 | 48.98 (unstable) | –0.107 (hydrophilic) |
| WP_006011447.1(WbkB) | 284 | 32425.00 | 5.52 | 49.28 (unstable) | –0.090 (hydrophilic) |
| WP_006642693.1(WbkA) | 376 | 42431.96 | 9.36 | 35.57 (stable) | –0.089 (hydrophilic) |
| WP_280176887.1 (WbkA) | 376 | 42419.91 | 9.36 | 33.66 (stable) | –0.103 (hydrophilic) |
| WP_002969840.1 (WbkA) | 376 | 42446.03 | 9.42 | 35.45 (stable) | –0.090 (hydrophilic) |
| WP_004683864.1 (FabZ) | 157 | 17186.95 | 5.60 | 26.64 (stable) | 0.088 (hydrophobic) |
| WP_004688426.1 (FabZ) | 157 | 17187.94 | 5.78 | 25.41 (stable) | 0.059 (hydrophobic) |
| WP_002964280.1 (FabZ) | 157 | 17213.97 | 5.60 | 25.41 (stable) | 0.071 (hydrophobic) |
| WP_004681750.1 (gmd) | 356 | 40681.44 | 5.71 | 33.05 (stable) | –0.545 (hydrophilic) |
| WP_004687379.1 (gmd) | 356 | 40667.42 | 5.71 | 32.82 (stable) | –0.544 (hydrophilic) |
| WP_006015606.1 (gmd) | 356 | 40731.52 | 5.71 | 33.64 (stable) | –0.541 (hydrophilic) |
| WP_002963680.1 (gmd) | 362 | 41062.55 | 5.98 | 32.55 (stable) | –0.395 (hydrophilic) |
| WP_004684016.1 (lpxC) | 286 | 30866.93 | 4.94 | 33.21 (stable) | 0.013 (hydrophilic) |
| WP_002964532.1 (lpxC) | 286 | 30852.91 | 4.94 | 33.08 (stable) | 0.012 (hydrophilic) |
Mw = Molecular weight, aa = Amino acid, pI = Isoelectric point, GRAVY = Grand average of hydropathicity
Prediction of subcellular localization
Using the DeepLocPro 1.0 server, prokaryotic proteins can be assigned to six localities: cytoplasm, cytoplasmic membrane, periplasm, outer membrane, cell wall and surface, and extracellular space. The potential subcellular location predictions of virulence proteins were obtained for lpxC, gmd, FabZ, WbkA, and WbkC as cytoplasmic, WbkB, and VirB3 as cytoplasmic membrane, VirB7 as outer membrane, and VirB5 as extracellular Table 4.
Table 4. Prediction of the probabilities of subcellular locations for virulence proteins in Brucella sp.
| Protein | Localization | Cell wall and surface | Extracelular | Cytoplasmic | Cytoplasmic membrane | Outer membrane | Periplasmic |
|---|---|---|---|---|---|---|---|
| VirB3 | Cytoplasmic Membrane | 0.0003 | 0.0023 | 0.0172 | 0.8849* | 0.0935 | 0.0018 |
| VirB7 | Outer Membrane | 0.0001 | 0.0019 | 0.0005 | 0.0652 | 0.9315* | 0.0008 |
| VirB5 | Extracellular | 0.0008 | 0.8865* | 0.0007 | 0.0019 | 0.0879 | 0.0222 |
| WbkC | Cytoplasmic | 0.0000 | 0.0014 | 0.9810* | 0.0077 | 0.0098 | 0.0001 |
| WbkB | Cytoplasmic Membrane | 0.0009 | 0.0288 | 0.2660 | 0.6487* | 0.0517 | 0.0039 |
| WbkA | Cytoplasmic | 0.0005 | 0.0036 | 0.7018* | 0.2400 | 0.0535 | 0.0006 |
| FabZ | Cytoplasmic | 0.0000 | 0.0001 | 0.9864* | 0.0090 | 0.0042 | 0.0003 |
| gmd | Cytoplasmic | 0.0004 | 0.0062 | 0.9327* | 0.0186 | 0.0313 | 0.0107 |
| lpxC | Cytoplasmic | 0.0000 | 0.0006 | 0.9749* | 0.0003 | 0.0239 | 0.0002 |
Higher subcellular location scores are indicated by asterisks.
Conserved domain identification and motif analysis
Information on conserved domains of virulence proteins was obtained from the CDD at NCBI and is presented in Table 5. The online MEME server was used to identify conserved motifs for the VirB3, VirB7, VirB5, WbkC, WbkB, WbkA, FabZ, gmd, and lpxC proteins and showed Table 6 and Figure 2.

Figure 2. Schematic representation of the conserved motif of virulence proteins of Brucella melitensis bv. 1 str. 16M, Brucella suis 1330, Brucella canis ATCC 23365 and Brucella abortus 2308.
Table 5. Conserved domain of virulence proteins in Brucella spp.
| Protein | Domain name | Accession | Description | Interval | E-value |
|---|---|---|---|---|---|
| VirB3 | VirB3 | COG3702 | Type IV secretory pathway, VirB3 component | 18-113 | 1.65 × 10-31 |
| VirB7 | YcfL | COG5633 | Uncharacterized conserved protein YcfL | 1-43 | 3.59 × 10-5 |
| VirB5 | VirB5 | TIGR02791 | P-type DNA transfer protein VirB5 | 1-217 | 4.42 × 10-98 |
| WbkC | FMT_core | cd08369 | Formyltransferase, catalytic core domain | 9-182 | 4.90 × 10-62 |
| WbkB | No domains identified | ||||
| WbkA | GT4_MtfB like | cd03809 | glycosyltransferases MtfB, WbpX, and similar proteins | 6-362 | 2.65 × 10-100 |
| FabZ | fabZ | PRK00006 | 3-hydroxyacyl-ACP dehydratase FabZ | 6-150 | 7.22 × 10-86 |
| gmd | Gmd | COG1089 | GDP-D-mannose dehydratase [Cell wall/membrane/envelope biogenesis] | 4-343 | 0 |
| lpxC | LpxC | COG0774 | UDP-3-O-acyl-N-acetylglucosamine deacetylase [Cell wall/membrane/envelope biogenesis] | 4-279 | 2.97 × 10-157 |
Table 6. The identified motif of virulence proteins in Brucella spp.
| Motif | Symbol | E-value | Sites | Width | Motif Consensus |
|---|---|---|---|---|---|
| 1 | red | 2.2e-098 | 20 | 20 | YNSIIEIDPRFIREAEVVLP |
| 2 | blue | 2.4e-100 | 22 | 21 | YSNLPHNYRDLYEAVMSGGIL |
| 3 | green | 4.0e-094 | 8 | 29 | MKKIILSFAFALTVTSTAHAQLPVTDAGS |
Multiple sequence alignment and phylogenetic analysis
When the resulting VirB3, VirB7, VirB5, WbkC, WbkB, WbkA, FabZ, gmd, and lpxC proteins multiple sequences alignment was visualized in Geneious Prime version 2024.0 (Figure 3) conserved and distinct amino acids were identified in the B. melitensis bv. 1 str. 16M, B. suis 1330, B. canis ATCC 23365, B. ovis ATCC 25840 and B. abortus 2308 virulence proteins sequences. The evolutionary history was inferred by using the Maximum Likelihood method and Jones–Taylor–Thornton (JTT) matrix-based model [22]. The tree with the highest log likelihood (-6454.69) is shown. The percentage of trees in which the associated taxa clustered together is shown above the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with the superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 24 amino acid sequences. There were 424 positions in the final dataset. Evolutionary analyses were conducted in MEGA11 [23] (Figure 4).
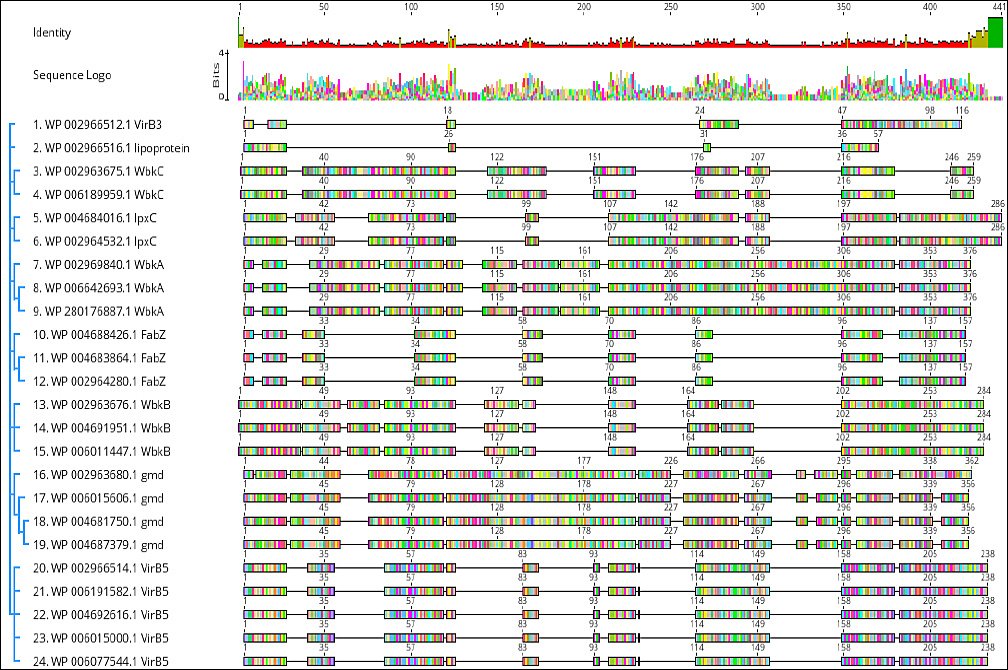
Figure 3. Multiple sequence alignment of VirB3, VirB7, VirB5, WbkC, WbkB, WbkA, FabZ, gmd, and lpxC proteins of Brucella melitensis bv. 1 str. 16M, Brucella suis 1330, Brucella canis ATCC 23365, Brucella ovis ATCC 25840 and Brucella abortus 2308. Geneious version 2024.0 created by Biomatters. Available from https://www.geneious.com.
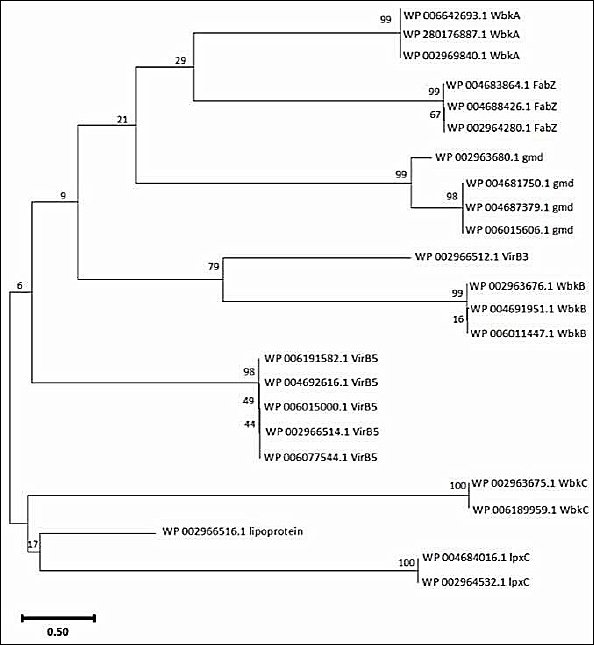
Figure 4. Phylogenetic analysis of virulence proteins of Brucella sp. The phylogenetic tree was generated using the amino acid sequences of selected virulence proteins by applying the Maximum Likelihood method and the JTT matrix-based model.
Interactive analysis of virulence proteins of B. abortus 2308
The interactive protein study unfolds the numerous interacting partners of VirB3, WbkB, WbkC, FabZ, gmd, and lpxC (Figure 5) and their interactive protein partners (Table 7). The VirB2-4-5-6-8-9-10-11 had very high interactions with reference protein VirB3 (Figure 5a). The reference VirB3 protein was also highly associated with the VirB1 and VirB7. The BAB1_0999, BAB1_0058, BAB1_0542, RfbD, gmd, BAB1_0544, BAB1_0561, and BAB2_0856 had high interactions with reference protein WbkB (BAB1_0541) (Figure 5b). BAB1_0999 is a conserved hypothetical protein that shows glycosyltransferase activity and functions in the LPS biosynthetic process. The activities of BAB1_0058, BAB1_0542, RfbD, gmd, BAB1_0544, BAB1_0561, and BAB2_0856 proteins are summarized in Table 7. In addition, BAB1_0802 ve BAB1_0540 had medium interactions with reference protein WbkB [24, 25, 26]. The BAB2_0695 had very high interactions with reference protein WbkC (BAB1_0540) (Figure 5c). BAB2_0695 has NAD-dependent epimerase/dehydratase activity [24]. NusB, BAB1_0544, FolD, Sun, BAB2_0845, BAB1_0188, BAB1_0542, RfbD, whose activities are summarized in Table 7, had high interactions with reference protein WbkC. The lpxA, lpxD, AcpP, AccA, FabD, AccD, lpxC, FabB, FabH, and BAB1_0486 had very high interactions with reference protein FabZ (Figure 5d). The BAB1_0802 had a very high interaction with the reference protein gmd (Figure 5e). Also, BAB1_0544, BAB1_0542, BAB2_0856, BAB1_0561, RfbD, BAB1_0541, Apt, RplD, and BAB1_0534 had high interactions with the reference protein gmd. The lpxA, lpxD, FabZ, lpxB, lpxK, and KdsA had very high interactions with the reference protein lpxC (Figure 5f). Also, the KdtA, KdsB, FtsZ, and SecA had high interactions with the reference protein lpxC.
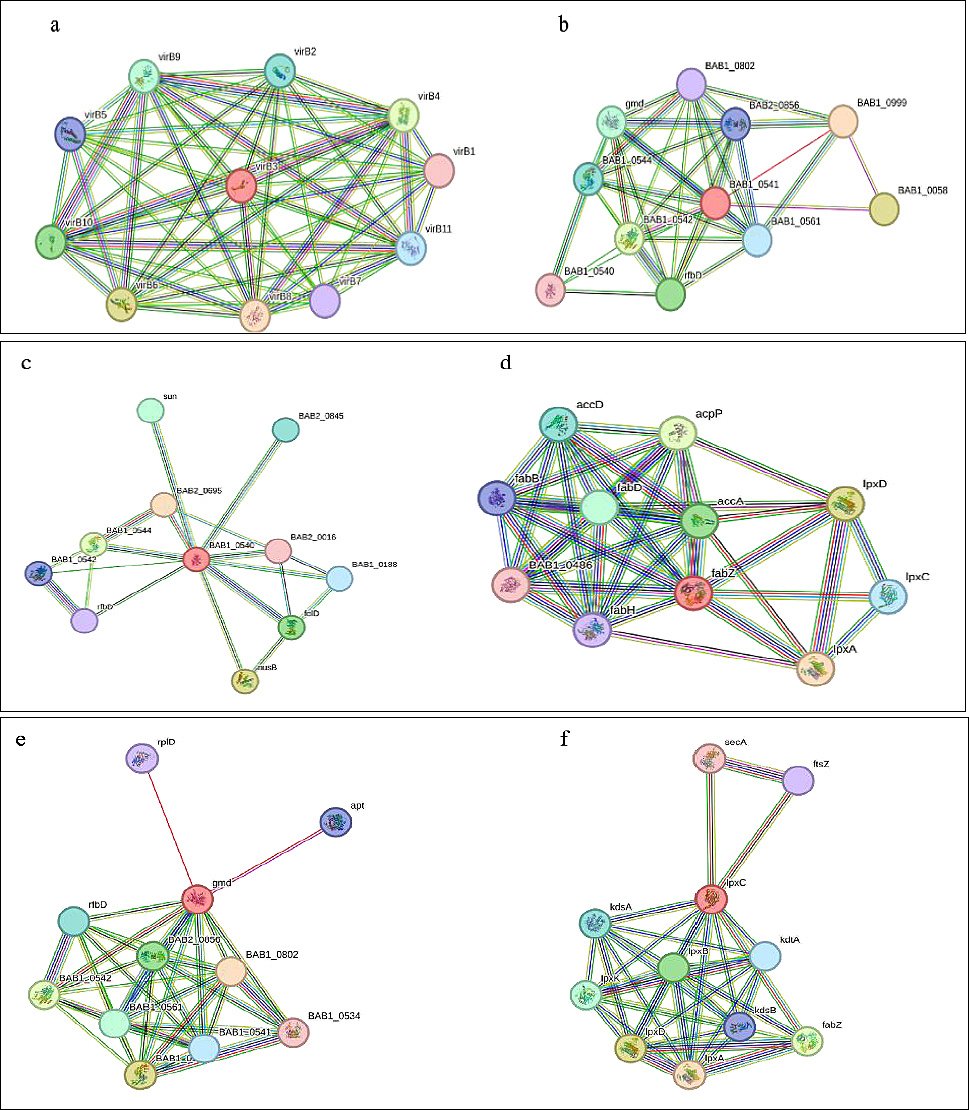
Figure 5. Interactive network of virulence proteins of Brucella abortus 2308. a) Interactive network of virulence protein VirB3, b) Interactive network of virulence protein WbkB, c) Interactive network of virulence protein WbkC, d) Interactive network of virulence protein FabZ, e) Interactive network of virulence protein gmd, and f) Interactive network of virulence protein lpxC.
Table 7. Interactive network of virulence proteins of Brucella abortus 2308.
| Virulence protein | Predicted functional partners name | Predicted functional partners |
|---|---|---|
| VirB3 | VirB8 | Type IV secretion system protein |
| VirB6 | Type IV secretion system protein | |
| VirB4 | Type IV secretion system protein | |
| VirB10 | Glutelin: Proline-rich region:Bacterial conjugation TrbI-like protein; | |
| VirB9 | Type IV secretion system CagX conjugation protein | |
| VirB2 | K03197 type IV secretion system protein | |
| VirB11 | Type IV secretion system protein | |
| VirB5 | Attachment mediating protein | |
| VirB7 | Type IV secretion system protein | |
| VirB1 | SLT domain | |
| WbkB | BAB1_0999 | Conserved hypothetical protein |
| BAB1_0058 | Esterase/lipase/thioesterase, active site | |
| BAB1_0542 | ATP/GTP-binding site motif A (P-loop):ABC transporter:AAA ATPase. | |
| RfbD | ABC transporter, family 2: Acriflavin resistance protein. | |
| gmd | Short-chain dehydrogenase/reductase SDR | |
| BAB1_0544 | DegT/DnrJ/EryC1/StrS aminotransferase | |
| BAB1_0561 | Mannose-6-phosphate isomerase, type II | |
| BAB2_0856 | Mannose-6-phosphate isomerase, type II | |
| BAB1_0802 | Bacterial sugar transferase. | |
| BAB1_0540 | Formyl transferase, N-terminal. | |
| WbkC | BAB2_0695 | NAD-dependent epimerase/dehydratase. |
| NusB | Antitermination protein | |
| BAB1_0544 | DegT/DnrJ/EryC1/StrS aminotransferase. | |
| FolD | Tetrahydrofolate dehydrogenase/cyclohydrolase | |
| Sun | SAM (and some other nucleotide) binding motif | |
| BAB2_0845 | SAM (and some other nucleotide) binding motif | |
| BAB1_0188 | Methionine synthase | |
| BAB1_0542 | ATP/GTP-binding site motif A (P-loop) | |
| RrfbD | ABC transporter, family 2 | |
| BAB2_0016 | Condensation domain. | |
| FabZ | lpxA | Bacterial transferase hexapeptide repeat. |
| lpxD | UDP-3-O-acylglucosamine N-acyltransferase. | |
| AcpP | Acyl carrier protein (ACP). | |
| AccA | Acetyl-CoA carboxylase, alpha subunit. | |
| FabD | Acyl transferase domain. | |
| AccD | Acetyl-CoA carboxylase carboxyl transferase, beta subunit. | |
| lpxC | UDP-3-0-acyl N-acetylglucosamine deacetylase. | |
| FabB | Beta-ketoacyl synthase. | |
| FabH | β-ketoacyl-ACP synthase III | |
| BAB1_0486 | Protein kinase: Beta-ketoacyl synthase. | |
| gmd | BAB1_0802 | Bacterial sugar transferase. |
| BAB1_0544 | DegT/DnrJ/EryC1/StrS aminotransferase. | |
| BAB1_0542 | ATP/GTP-binding site motif A (P-loop). | |
| BAB2_0856 | Mannose-6-phosphate isomerase, type II. | |
| BAB1_0561 | Mannose-6-phosphate isomerase, type II. | |
| RfbD | ABC transporter, family 2. | |
| BAB1_0541 | Perosamine synthetase WbkB. | |
| Apt | Adenine phosphoribosyltransferase. | |
| RplD | Ribosomal protein L4/L1e. | |
| BAB1_0534 | Polysaccharide biosynthesis protein CapD. | |
| lpxC | lpxA | Bacterial transferase hexapeptide repeat. |
| lpxD | UDP-3-O-acylglucosamine N-acyltransferase. | |
| FabZ | Thioesterase superfamily. | |
| lpxB | Glycosyl transferase, family 19. | |
| lpxK | Tetraacyldisaccharide-1-P 4’-kinase. | |
| KdsA | DAHP synthetase I/KdsA superfamily. | |
| KdtA | Three-deoxy-D-manno-octulosonic-acid transferase, N-terminal. | |
| KdsB | Acylneuraminate cytidylyltransferase. | |
| FtsZ | Cell division protein FtsZ. | |
| SecA | SecA protein. |
ABC = ATP-binding cassette, ATP/GTP = Adenosine Triphosphate/Guanosine Triphosphate, DAHP = 3-Deoxy-D-arabinoheptulosonate 7-phosphate, NAD = Nicotinamide Adenine Dinucleotide, SAM = S-Adenosyl methionine, SDR = The short-chain dehydrogenases/reductase, SLT = Soluble Lytic Transglycosylases, UDP-3-O-acylglucosamine = Uridine diphosphate-3-O-acylglucosamine
DISCUSSION
Physicochemical characteristics of selected virulence proteins
Brucella species are facultative intracellular pathogens known to cause serious zoonotic diseases around the world [27]. The virulence of these pathogens depends on their ability to evade or modulate the host immune response, as well as their capacity to survive and replicate within a vacuole derived from the endoplasmic reticulum [28].
Proteins with an instability index above 40 indicate that the protein is unstable and has a short half-life [29]. The GRAVY value is commonly used to evaluate the hydrophilicity and hydrophobicity of proteins. Moreover, the GRAVY value ranges between −2 and 2, where a negative or positive value indicates that the protein possesses hydrophilic or hydrophobic properties, respectively [30]. VirB3, VirB7, and FabZ proteins are predicted to be hydrophobic, whereas VirB5, WbkC, WbkB, WbkA, gmd, and lpxC proteins are predicted to be hydrophilic.
Conserved domains and motif architecture of virulence-associated proteins
Considering the properties of the conserved domains of VirB3, VirB7, VirB5, WbkC, WbkA, FabZ, gmd, and lpxC proteins, respectively, the VirB3 protein contains the VirB3 superfamily covering amino acids 18-113, the VirB7 protein contains the putative periplasmic lipoprotein domain between amino acids 1-43, and the VirB5 protein contains the VirB5_like domain covering amino acids 1-217 [31]. There are main genes in the wbk locus, and one of the proteins encoded by these genes, the WbkC protein, contains the formyltransferase catalytic core domain encompassing amino acids 9-182 [17]. When the conserved domain for the WbkA protein was examined, it was observed that it belongs to glycosyltransferase family 4 (GT4) between amino acids 6-362 [32]. The gmd protein contains the GDP-D-mannose dehydratase domain between amino acids 4-343 [33]. The FabZ protein contains the 3-hydroxyacyl-ACP dehydratase FabZ domain between amino acids 6-150 [24]. The lpxC protein contains the UDP-3-O-acyl-N-acetylglucosamine deacetylase domain between amino acids 4-279.
Protein sequence motifs are patterns of residues that exhibit distinct structural and functional characteristics. They have been identified using multiple sequence and structural alignment methods as conserved signatures typical of protein families [34]. An important aspect of biological sequence characterization is the identification of motifs and domains [35]. Three conserved motifs were obtained:”YNSIIEIDPRFIREAEVVLPV” (Motif 1, red), “YSNLPHNYRDLYEAVMSGGIL” (Motif 2, blue), and “MKKIILSFAFALTVTSTAHAQLPVTDAGS” (Motif 3, green). B. melitensis bv. 1 str. 16M, B. suis 1330, B. canis ATCC 23365, B. ovis ATCC 25840, and B. abortus 2308 had all three motifs in VirB5 and WbkB virulence proteins. Motif 1 (red) and Motif 2 (blue) were detected in WbkA, FabZ, gmd, and lpxC virulence proteins. No motif was detected in VirB3 and VirB7 virulence proteins. All virulence proteins except VirB3, VirB7, and WbkC have the “YNSIIEIDPRFIREAEVVLPV” motif represented in Motif 1 (red). The conserved motif analysis and the phylogenetic tree are consistent with each other.
Functional significance of protein–protein interaction networks
Understanding biological processes requires a well-organized understanding of the physical interactions between proteins [36]. Proteins are essential molecules in living organisms, performing a variety of critical functions within cells, such as signaling, regulating metabolism, and maintaining cell structure [37]. Proteins interact with one another, regulating each other’s functions and binding together to fulfill their roles in biological processes [36]. Investigating protein–protein interactions not only enhances our understanding of vital cellular functions but also plays a significant role in the development of disease treatments and the design of drugs [37].
Virulence factors are essential for pathogen infection, making the interaction of virulence factors particularly important. LPS is an essential virulence factor of Brucella [5]. The virulence factors lpxA, lpxC, KdsB, and KdsA are involved in different stages of lipid A biosynthesis, an essential component of the LPS structure [38].
lpxC as a potential antimicrobial target in Brucella
Although lpxC has been extensively validated as a drug target in other Gram-negative pathogens such as E. coli, Pseudomonas aeruginosa, and Acinetobacter baumannii [39, 40], its role as a therapeutic target in Brucella has not been widely addressed. Protein-protein interaction analyses have revealed that lpxC interacts significantly with a variety of proteins. Inhibition of lpxC may influence multiple interconnected components of the LPS biosynthetic pathway rather than definitively destabilizing the entire machinery. To the best of our knowledge, lpxC has not been widely emphasized as a rational drug target in Brucella, and our in silico network, motif, and localization data provide a complementary perspective for anti-Brucella drug development.
Vaccine relevance of type IV secretion system-associated proteins
The interactive network analysis in this study highlights that inhibiting one of the proteins such as VirB3, VirB5, VirB7, WbkC, WbkA, FabZ, gmd, or lpxC may affect several functionally associated proteins and biological processes rather than guaranteeing a direct cascade failure. This synergistic vulnerability suggests that these proteins are not only potential drug targets but may also represent promising vaccine candidates when surface-exposed or secreted (e.g., VirB5, VirB7). Moreover, the conserved motifs identified, particularly those shared across Brucella species, may provide a basis for designing multi-epitope subunit vaccines aimed at inducing broad protective immunity [19, 41–44].
Brucella T4SS proteins play critical roles in the intracellular life cycle of the bacterium and may serve as targets for the immune system. In the literature, studies on VirB proteins have mostly focused on single antigens. For example, Tan et al. [45] reported that recombinant VirB5 protein could serve as a strong antigen for the serological diagnosis of bovine brucellosis, demonstrating high specificity (97.8%). Similarly, Pollak et al. [46] showed that immunization with VirB7 in combination with VirB9 in mice and dogs elicited a Th1-type immune response and reduced splenic bacterial load by approximately 1 log.
Comparative significance of VirB3, VirB5, and VirB7 in target prioritization
VirB3 is located in the cytoplasmic membrane, which limits its immune accessibility. The importance of VirB3 does not stem from sequence conservation or surface exposure, but rather from its central functional role within the T4SS and its strong connections with multiple VirB components in the protein–protein interaction network. This network centrality supports prioritizing VirB3 as a functional drug target rather than as a primary vaccine candidate.
By comparison, although VirB5 and VirB7 are not absolute central hubs in the interaction network, they are functionally associated with the T4SS and show significant interactions with VirB3, indicating biological relevance within the system. When their immune accessibility advantage is considered, these two proteins emerge as complementary and potentially synergistic candidates for subunit vaccine design.
Broad-spectrum vaccine implications of the VirB5-VirB7 combination
Our findings indicate that the combined evaluation of VirB5 and VirB7 may provide a rational basis for a potential broad-spectrum subunit vaccine perspective. VirB5 was shown to harbor conserved motifs across all Brucella species in this study and to be extracellularly localized, while VirB7 was predicted to be an outer membrane-associated lipoprotein with strong interactions within the T4SS network. These features suggest that both proteins could represent promising core immunogens for a multi-epitope vaccine strategy rather than isolated antigen selection.
Nevertheless, considering the motif distribution observed in our study, the likelihood of stronger cross-protective potential appears particularly high for five Brucella spp., where VirB5 motifs were consistently conserved. These conclusions are consistent with previous reports highlighting conserved outer membrane and T4SS proteins as cross-protective antigens and align with recent reviews emphasizing multi-antigen subunit or motif-based vaccines as promising next-generation strategies [19, 20, 41–44, 46, 47].
To the best of our knowledge, the VirB5-VirB7 combination has received limited attention in previous Brucella vaccine studies, and our findings support its consideration as a potential vaccine target. It should be noted that the distinct contribution of the present study does not arise from the individual bioinformatic tools employed, which are widely used in molecular analyses, but from their integrative interpretation across multiple complementary analytical dimensions. This combined evaluation enabled a comparative prioritization of conserved virulence factors rather than a purely descriptive characterization, thereby providing a more rational and evidence-guided framework for translational vaccine and drug target exploration.
One Health relevance of conserved virulence factors across Brucella species
Since the Brucella species included in this study are known to infect different hosts, the conserved proteins identified here may have relevance across multiple species rather than being limited to a single host. From a One Health perspective, recognizing shared virulence mechanisms between animal and human infections can contribute to more integrated prevention and control strategies. Although these findings are based on computational analyses, the cross-species conservation observed suggests a possible value for broader zoonotic surveillance, vaccine research, and antimicrobial target exploration in both veterinary and human health contexts.
CONCLUSION
This study provides a comprehensive in silico evaluation of key virulence-associated proteins (VirB3, VirB5, VirB7, WbkC, WbkB, WbkA, FabZ, gmd, and lpxC) across multiple Brucella species, highlighting their conserved structural, physicochemical, and functional characteristics. The results demonstrated that several proteins, particularly VirB5, VirB7, WbkA, FabZ, gmd, and lpxC, exhibit favorable stability profiles and conserved domains associated with essential biological functions such as type IV secretion system activity, LPS biosynthesis, and intracellular survival. Motif analysis revealed shared conserved sequences across species, supporting their evolutionary conservation and potential functional importance. Additionally, protein–protein interaction network analysis identified strong connectivity among virulence factors, indicating their coordinated role in pathogenicity.
From a practical perspective, these findings suggest that selected virulence proteins may serve as promising candidates for the development of novel diagnostic, therapeutic, and preventive strategies. In particular, lpxC emerges as a potential antimicrobial target due to its central role in lipid A biosynthesis and its extensive interaction network. Similarly, extracellular and outer membrane-associated proteins such as VirB5 and VirB7 demonstrate strong potential as vaccine candidates due to their accessibility to the host immune system with VirB5 further distinguished by its conserved motif architecture, which supports its potential as a promising vaccine target.. The identification of shared motifs further supports the feasibility of designing multi-epitope or subunit vaccines with broader cross-protective potential across Brucella species.
A major strength of this study lies in the integrative analytical approach, combining physicochemical characterization, conserved domain identification, motif analysis, subcellular localization prediction, phylogenetic relationships, and protein–protein interaction networks. This multi-dimensional framework enables a more rational prioritization of virulence factors compared to single-method analyses. Furthermore, the inclusion of multiple Brucella species representing diverse hosts enhances the translational relevance of the findings within a One Health context.
However, this study has several limitations. The conclusions are based solely on computational predictions and database-derived sequences, which may not fully reflect biological complexity under in vivo conditions. Functional validation of the identified targets, including gene knockout studies, protein expression analyses, and immunogenicity assessments, was not performed. In addition, host-specific immune responses and environmental factors influencing protein expression were not considered.
Future research should focus on experimental validation of the prioritized targets, particularly lpxC, VirB5, and VirB7, using in vitro and in vivo models. Structural biology approaches, such as protein crystallography and molecular docking studies, may further clarify their suitability as drug targets. Moreover, vaccine development studies incorporating conserved motifs into multi-epitope constructs should be explored to assess cross-protective immunity across different Brucella species. Integrating genomic, proteomic, and immunological data will further strengthen the translational applicability of these findings.
In conclusion, this study provides a systematic and integrative framework for identifying conserved virulence-associated proteins in Brucella spp. and highlights their potential relevance in antimicrobial and vaccine development. The findings contribute to a deeper understanding of Brucella pathogenesis and support the advancement of targeted control strategies within a One Health perspective.
DATA AVAILABILITY
The supplementary data can be available from the corresponding author upon a reasonable request. Figure S1-S9: Prediction of subcellular localization of virulence protein of Brucella spp.; Table S1: Motif analysis parameters of virulence protein of Brucella spp.; Figure S10: Multiple sequence alignment of virulence protein of Brucella spp.; Protein sequences of virulence proteins of Brucella spp.; Table S2-7: Node degree of virulence proteins of VirB3, WbkB, WbkC, FabZ, gmd, and lpxC; Figure S11-16: Predicted functional partners of VirB3, WbkB, WbkC, FabZ, gmd, and lpxC; Figure S17: STRING Settings parameters.
AUTHORS’ CONTRIBUTIONS
AO and SY: Conceptualization, investigation, writing – original draft, validation, writing – review and editing. Both authors have read and approved the final manuscript.
COMPETING INTERESTS
The authors declare that they have no competing interests.
PUBLISHER’S NOTE
Veterinary World remains neutral with regard to jurisdictional claims in the published institutional affiliations.
ACKNOWLEDGMENTS
The authors did not receive any funds for this study. The authors are thankful to Istanbul Gelişim University and Mugla Sitki Kocman University for providing the necessary facilities for this study.
REFERENCES
- Pal M, Kerorsa GB, Desalegn C, Kandi V. Human and animal brucellosis:a comprehensive review of biology, pathogenesis, epidemiology, risk factors, clinical signs, laboratory diagnosis. Am J Infect Dis 2020;8:118-126. [Google Scholar] | [Crossref]
- Byndloss MX, Tsolis RM. Brucella spp. virulence factors and immunity. Annu Rev Anim Biosci 2016;4(1):111-127. [Google Scholar] | [Crossref]
- Kamga RM, Silatsa BA, Farikou O, Kuiate JR, Simo G. Detection of Brucella antibodies in domestic animals of southern Cameroon:implications for the control of brucellosis. Vet Med Sci 2020;6:410-420. [Google Scholar] | [Crossref]
- González-Espinoza G, Arce-Gorvel V, Mémet S, Gorvel JP. Brucella:reservoirs and niches in animals and humans. Pathogens 2021;10(2):186. [Google Scholar] | [Crossref]
- Głowacka P, Żakowska D, Naylor K, Niemcewicz M, Bielawska-Drozd A. Brucella–virulence factors, pathogenesis and treatment. Pol J Microbiol 2018;67:151-161. [Google Scholar] | [Crossref]
- Khurana SK, Sehrawat A, Tiwari R, Prasad M, Gulati B, Shabbir MZ. Bovine brucellosis–a comprehensive review. Vet Q 2021;41:61-88. [Google Scholar] | [Crossref]
- Riegert AS, Chantigian DP, Thoden JB, Tipton PA, Holden HM. Biochemical characterization of WbkC, an N-formyltransferase from Brucella melitensis. Biochemistry 2017;56:3657-3668. [Google Scholar] | [Crossref]
- Zygmunt MS, Blasco JM, Letesson JJ, Cloeckaert A, Moriyón I. DNA polymorphism analysis of Brucella lipopolysaccharide genes reveals marked differences in O-polysaccharide biosynthetic genes between smooth and rough Brucella species and novel species-specific markers. BMC Microbiol 2009;9:92. [Google Scholar] | [Crossref]
- Tsvetkov YE, Volkov TM, Eremin SA, Sklyarov OD, Kulakov YK, Krylov VB. New synthesis of oligosaccharides modelling the M epitope of the Brucella O-polysaccharide. Front Chem 2024;1:1424157. [Google Scholar] | [Crossref]
- Lapaque N, Moriyón I, Moreno E, Gorvel JP. Brucella lipopolysaccharide acts as a virulence factor. Curr Opin Microbiol 2005;8:60-66. [Google Scholar] | [Crossref]
- Wattam AR, Inzana TJ, Williams KP, Mane SP, Shukla M, Almeida NF. Comparative genomics of early-diverging Brucella strains reveals a novel lipopolysaccharide biosynthesis pathway. mBio 2012;3(5):e00246-12. [Google Scholar] | [Crossref]
- Kornspan D, Lubkovskaia R, Mathur S, Yeheskel A, Salmon-Divon M. Genomic analysis of natural rough Brucella melitensis Rev.1 vaccine strains:identification and characterization of mutations in key genes associated with bacterial LPS biosynthesis and virulence. Int J Mol Sci 2020;21:9341. [Google Scholar] | [Crossref]
- Zhang L, Liu W, Hu T, Du L, Luo C, Chen K. Structural basis for catalytic and inhibitory mechanisms of β-hydroxyacyl-acyl carrier protein dehydratase (FabZ). J Biol Chem 2008;283:5370-5379. [Google Scholar] | [Crossref]
- Sankarasubramanian J, Vishnu US, Sridhar J, Gunasekaran P, Rajendhran J. Pan-genome of Brucella species. Indian J Microbiol 2015;55:88-101. [Google Scholar] | [Crossref]
- Godfroid F, Cloeckaert A, Taminiau B, Danese I, Tibor A, De Bolle X. Genetic organisation of the lipopolysaccharide O-antigen biosynthesis region of Brucella melitensis 16M (wbk). Res Microbiol 2000;151:655-668. [Google Scholar] | [Crossref]
- Christopher S, Umapathy BL, Ravikumar KL. Brucellosis:review on the recent trends in pathogenicity and laboratory diagnosis. J Lab Physicians 2010;2:55-60. [Google Scholar] | [Crossref]
- Cardoso PG, Macedo GC, Azevedo V, Oliveira SC. Brucella spp. noncanonical LPS:structure, biosynthesis, and interaction with host immune system. Microb Cell Fact 2006;5:13. [Google Scholar] | [Crossref]
- Holzer K, Hoelzle LE, Wareth G. Genetic comparison of Brucella spp and Ochrobactrum spp. erroneously included into the genus Brucella confirms separate genera. Ger J Vet Res 2023;3:31-37. [Google Scholar] | [Crossref]
- Xiong X, Li B, Zhou Z, Gu G, Li M, Liu J. The VirB system plays a crucial role in Brucella intracellular infection. Int J Mol Sci 2021;22:13637. [Google Scholar] | [Crossref]
- Yin Z, Li M, Niu C, Yu M, Xie X, Haimiti G. Design of multi-epitope vaccine candidate against Brucella type IV secretion system (T4SS). PLoS One 2023;18(8):e0286358. [Google Scholar] | [Crossref]
- Hasan TH, Kadhum HA, Alasedi KK. Brucella spp. virulence factors. Pharm Res;2021(13):09752366. [Google Scholar] | [Crossref]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. CABIOS 1992;8:275-282. [Google Scholar] | [Crossref]
- Tamura K, Stecher G, Kumar S. MEGA11:Molecular Evolutionary Genetics Analysis version 11. Mol Biol Evol 2021;38:3022-3027. [Google Scholar] | [Crossref]
- Chain PS, Comerci DJ, Tolmasky ME, Larimer FW, Malfatti SA, Vergez LM. Whole-genome analyses of speciation events in pathogenic Brucellae. Infect Immun 2005;73:8353-8361. [Google Scholar] | [Crossref]
- Lamontagne J, Beland M, Forest A, Cote-Martin A, Nassif N, Tomaki F. Proteomics-based confirmation of protein expression and correction of annotation errors in the Brucella abortus genome. BMC Genomics 2010;11:300. [Google Scholar] | [Crossref]
- Suarez-Esquivel M, Ruiz-Villalobos N, Castillo-Zeledon A, Jimenez-Rojas C, Roop MII, Comerci DJ. Brucella abortus strain 2308 Wisconsin genome:importance of the definition of reference strains. Front Microbiol 2016;7:1557. [Google Scholar] | [Crossref]
- Ronneau S, Moussa S, Barbier T, Conde-Alvarez R, Zuniga-Ripa A, Moriyón I. Brucella, nitrogen and virulence. Crit Rev Microbiol 2016;42(4):507-525. [Google Scholar] | [Crossref]
- Martirosyan A, Moreno E, Gorvel JP. An evolutionary strategy for a stealthy intracellular Brucella pathogen. Immunol Rev 2011;240(1):211-234. [Google Scholar] | [Crossref]
- Sganzerla Martinez G, Dutt M, Kumar A, Kelvin DJ. Multiple Protein Profiler 1.0 (MPP):a webserver for predicting and visualizing physiochemical properties of proteins at the proteome level. Protein J 2024;43(4):711-717. [Google Scholar] | [Crossref]
- Zhao S, Pan F, Cai S, Yi J, Zhou L, Liu Z. Secrets behind protein sequences:unveiling the potential reasons for varying allergenicity caused by caseins from cows, goats, camels, and mares based on bioinformatics analyses. Int J Mol Sci 2023;24(3):2481. [Google Scholar] | [Crossref]
- Yuan Q, Carle A, Gao C, Sivanesan D, Aly KA, Höppner C. Identification of the VirB4-VirB8-VirB5-VirB2 pilus assembly sequence of type IV secretion systems. J Biol Chem 2005;280:26349-26359. [Google Scholar] | [Crossref]
- Coutinho PM, Deleury E, Davies GJ, Henrissat B. An evolving hierarchical family classification for glycosyltransferases. J Mol Biol 2003;328:307-317. [Google Scholar] | [Crossref]
- Cloeckaert A, Vizcaíno N, Paquet JY, Bowden RA, Elzer PH. Major outer membrane proteins of Brucella spp.:past, present and future. Vet Microbiol. 2002;90((1-4)):229-247. [Google Scholar] | [Crossref]
- Savojardo C, Martelli PL, Casadio R. Finding functional motifs in protein sequences with deep learning and natural language models. Curr Res Struct Biol 2023;5:102641. [Google Scholar] | [Crossref]
- Xiong J. Protein motifs and domain prediction. Cambridge: Cambridge University Press; 2006. p. 85-94. [Google Scholar]
- Szklarczyk D, Nastou K, Koutrouli M, Kirsch R, Mehryary F, Hachilif R. The STRING database in 2025:protein networks with directionality of regulation. Nucleic Acids Res 2025;53((D1)):D730-D737. [Google Scholar] | [Crossref]
- Hu J, Li Z, Rao B, Thafar MA, Arif M. Improving protein–protein interaction prediction using protein language model and protein network features. Anal Biochem 2024;693:115550. [Google Scholar] | [Crossref]
- Gupta SK, Gross R, Dandekar T. An antibiotic target ranking and prioritization pipeline combining sequence, structure and network-based approaches exemplified for Serratia marcescens. Gene 2016;591(1):268-278. [Google Scholar] | [Crossref]
- Barb AW, Jiang L, Raetz CRH, Zhou P. Structure of the deacetylase LpxC bound to the antibiotic CHIR-090:time-dependent inhibition and specificity in ligand binding. Proc Natl Acad Sci U S A 2007;104(47):18433-18438. [Google Scholar] | [Crossref]
- Caughlan RE, Jones AK, Delucia AM, Woods AL, Xie L, Ma B. Mechanisms decreasing in vitro susceptibility to the LpxC inhibitor CHIR-090 in the Gram-negative pathogen Pseudomonas aeruginosa. Antimicrob Agents Chemother 2012;56(1):17-27. [Google Scholar] | [Crossref]
- Cloeckaert A, Grayon M, Verger JM, Letesson JJ, Godfroid F. Conservation of seven genes involved in the biosynthesis of the lipopolysaccharide O-side chain in Brucella spp. Res Microbiol 2000;151:209-216. [Google Scholar] | [Crossref]
- Ke Y, Wang Y, Li W, Chen Z. Type IV secretion system of Brucella spp. and its effectors. Front Cell Infect Microbiol 2015;5:72. [Google Scholar] | [Crossref]
- Heidary M, Dashtbin S, Ghanavati R, Mahdizade Ari M, Bostanghadiri N, Darbandi A. Evaluation of brucellosis vaccines:a comprehensive review. Front Vet Sci 2022;9:925773. [Google Scholar] | [Crossref]
- Nandini P, Jakka P, Murugan S, Mazumdar V, Kumar D, Prakash R. Immuno-profiling of Brucella proteins for developing improved vaccines and DIVA capable serodiagnostic assays for brucellosis. Front Microbiol 2023;14:1253349. [Google Scholar] | [Crossref]
- Tan W, Wang XR, Nie Y, Wang C, Cheng LQ, Wang XC. Recombinant VirB5 protein as a potential serological marker for the diagnosis of bovine brucellosis. Mol Cell Probes;2012(26):127-131. [Google Scholar] | [Crossref]
- Pollak CN, Wanke MM, Estein SM, Delpino MV, Monachesi NE, Comerci EA. Immunization with Brucella VirB proteins reduce organ colonization in mice through a Th1-type immune response and elicits a similar immune response in dogs. Clin Vaccine Immunol 2015;22(3):274-281. [Google Scholar] | [Crossref]
- Blasco JM, Moreno E, Muñoz PM, Conde-Álvarez R, Moriyón I. A review of three decades of use of the cattle brucellosis rough vaccine Brucella abortus RB51:myths and facts. BMC Vet Res;2023(19):211. [Google Scholar] | [Crossref]